Hva er Aminoaciduria?

Aminosyre er en tilstand der det er for store mengder aminosyrer i urinen på grunn av genetiske defekter i aminosyremetabolismens veier. En mangel på et enzym som resulterer i en defekt i aminosyremetabolismen kalles primær aminosyre. Defekter hos molekylære transportører som er ansvarlige for transport og absorpsjon av en aminosyre er klassifisert som sekundær aminosyre. Begge typer aminosyrer kan være arvelige, for det meste i et autosomalt recessivt mønster, men noen kan bli ervervet sekundært til flere sykdommer som hyperparatyreoidisme, multippelt myelom, osteomalasi, rakitt, og viral hepatitt.
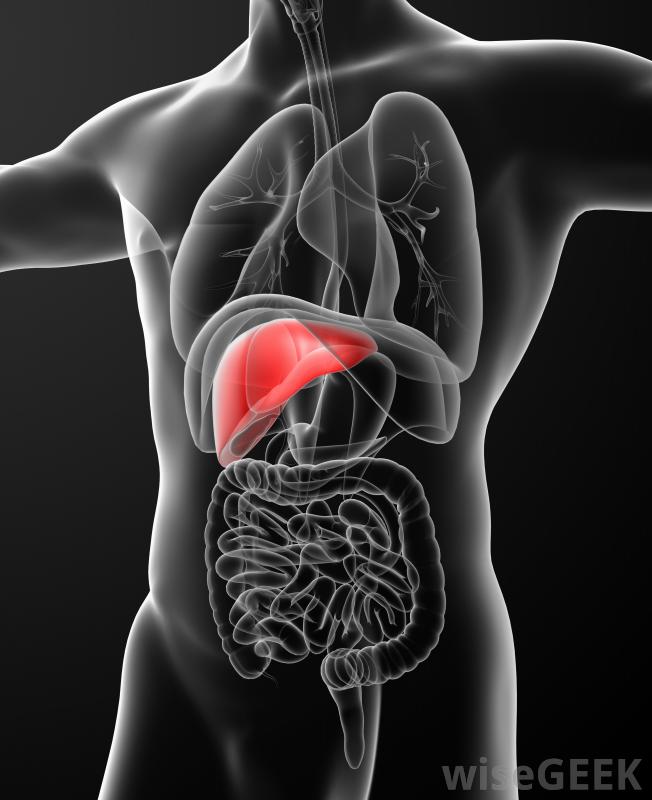
Noen vanlige eksempler på arvet primær aminosyre er klassisk fenylketonuri, klassisk homocystinuri, og alkaptonuri. Klassisk fenylketonuri er preget av en økt konsentrasjon av fenylalanin og dets biprodukter i vev, plasma, og urin på grunn av mangel på fenylalaninhydroksylase. De karakteristiske funnene ved klassisk fenylketonuri hos et ubehandlet barn inkluderer mental retardasjon, unnlatelse av å nå tidlige milepæler i utviklingen, mikrocefali, hypopigmentering av hud og hår, anfall, skjelving, hyperaktivitet, og unnlatelse av å vokse. Forebygging av disse funnene hos et barn kan gjøres ved tidlig diagnose og oppstart av kostholdsbehandling før 3 ukers alder.

Klassisk homocystinuri er preget av en økt konsentrasjon av homocystein og metionin, og redusert konsentrasjon av cystein i plasma og urin. Dette skyldes redusert aktivitet av cystathionine beta-synthase. Berørte individer som forskyver optiske linser kjent som ectopia lentis, mental retardasjon, skjelettavvik, osteoporose, og for tidlig arteriell sykdom. Behandlingen består av diettbegrensning av protein og metionin, og tilskudd med vitamin B 6 , B 12 , og folat.
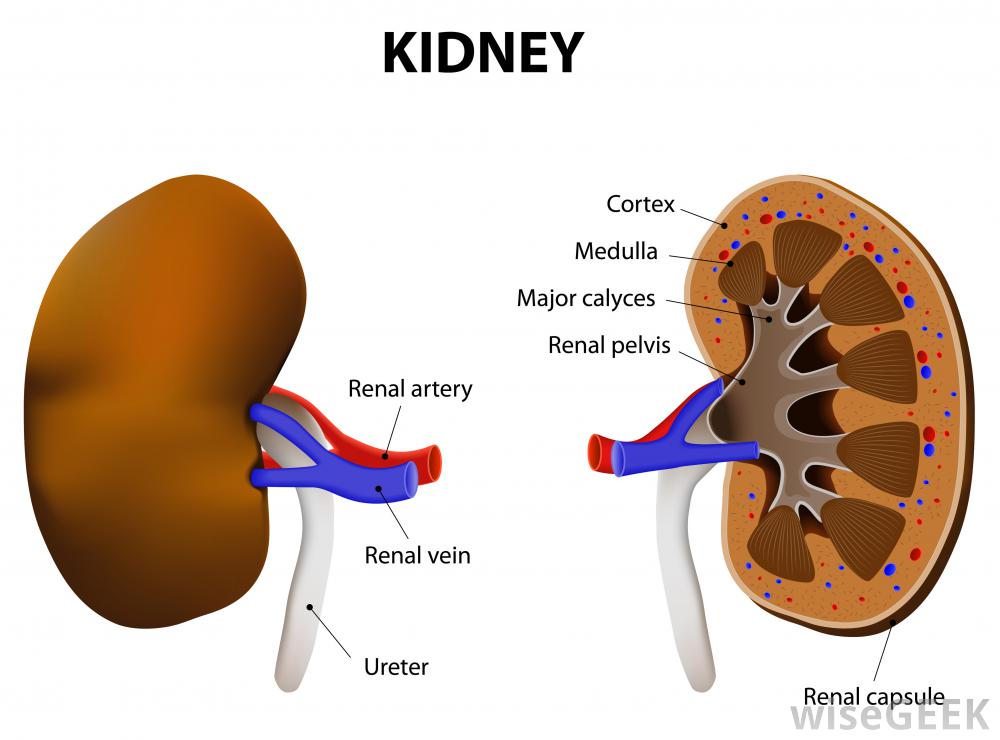
Alkaptonuria er preget av en økt konsentrasjon av homogentisinsyre i urin og bindevev på grunn av mangel på homogentisinsyreoksidase. Berørte individer er vanligvis asymptomatiske til de er i 30- eller 40 -årene. De tre karakteristiske symptomene på alkaptonuri er tilstedeværelsen av mørk urin, stor leddgikt, og mørkere ører og annet brusk og kollagen vev. Forebygging av langsiktige komplikasjoner kan gjøres gjennom begrensning av proteindiet, spesielt i fenylalanin og tyrosin, sammen med bruk av stoffet nitisone.
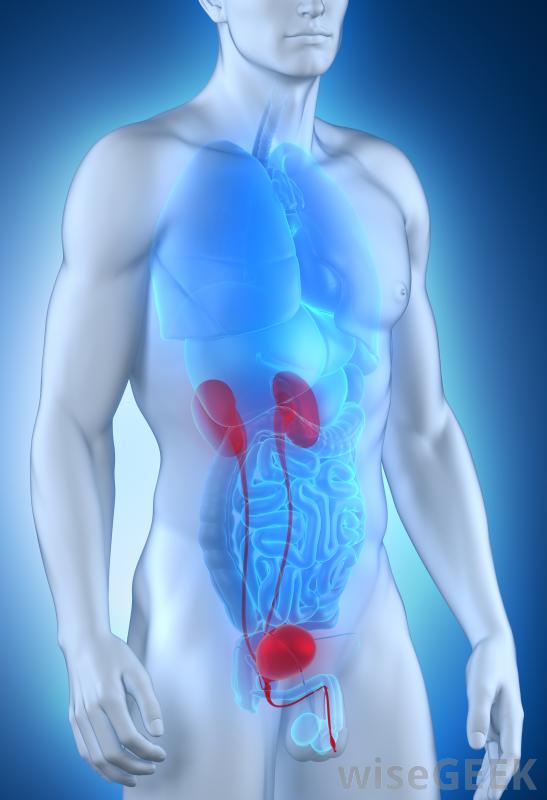
Noen vanlige eksempler på arvelig sekundær aminosyre er cystinuri, dibasisk aminosyre, og Hartnups sykdom. Cystinuri, på grunn av transportdefekt i nyre og tynntarm, er preget av nedsatt reabsorpsjon og overdreven utskillelse av dibasiske aminosyrer cystin, arginin, lysin, og ornitin i urinen. Dårlig løselighet av cystin disponerer for dannelse av nyre, urinleder, og blærestein, som kan føre til nyresvikt. Behandlingsmålet er å forhindre steindannelse ved livslang alkalisk diurese. Avhengig av presentasjonen av berørte personer, bruk av penicillamin og tiopronin, sjokkbølge litotripsy, ureteroskopi, perkutan nefrolitotomi, eller åpen urologisk kirurgi kan vurderes.
Dibasisk aminosyre er preget av en selektiv defekt i reabsorpsjonen av arginin, lysin, og ornitin. Berørte personer kan få en forstørret lever, proteinintoleranse, hyperammonemi, nedsatt nyrefunksjon, alvorlig osteoporose, eller strukturelle endringer i lungene. Behandlingen består av begrensninger i proteindiet og tilskudd av citrullin.
Hartnups sykdom er preget av varierende nevrologiske manifestasjoner, som cerebellar ataksi eller delirium, ledsaget av pellagra-lignende hudskader. Dette skyldes en defekt hos en transportør lokalisert i nyrer og tarm, resulterer i økt urinutskillelse av alanin, treonin, leucine, isoleucin, asparagine, glutamin, histidin, serine, tyrosin, valine, tryptofan, og fenylalanin. Behandlingen inkluderer en diett som inneholder mye proteiner og nikotinamid.